ePoster

QR unavailable
Share ePoster
Scan or copy the public World Wide URL.
ePoster
ANTIPRIONS: FUNCTIONAL VARIOMICS TO TACKLE NEUROTOXIC AMYLOID AGGREGATION
Blanca Poquet-Fullanaand 4 co-authors
Molecular Biology Institute of Barcelona (IBMB-CSIC)
FENS Forum 2026 (2026)
Barcelona, Spain
Presenter and authors
Presenter
Blanca Poquet-Fullana
Molecular Biology Institute of Barcelona (IBMB-CSIC)
Co-authors
Kimberly Alomoto-Balseca; Linda Luna-Iturra; Carme Gallego; Martí Aldea
Abstract
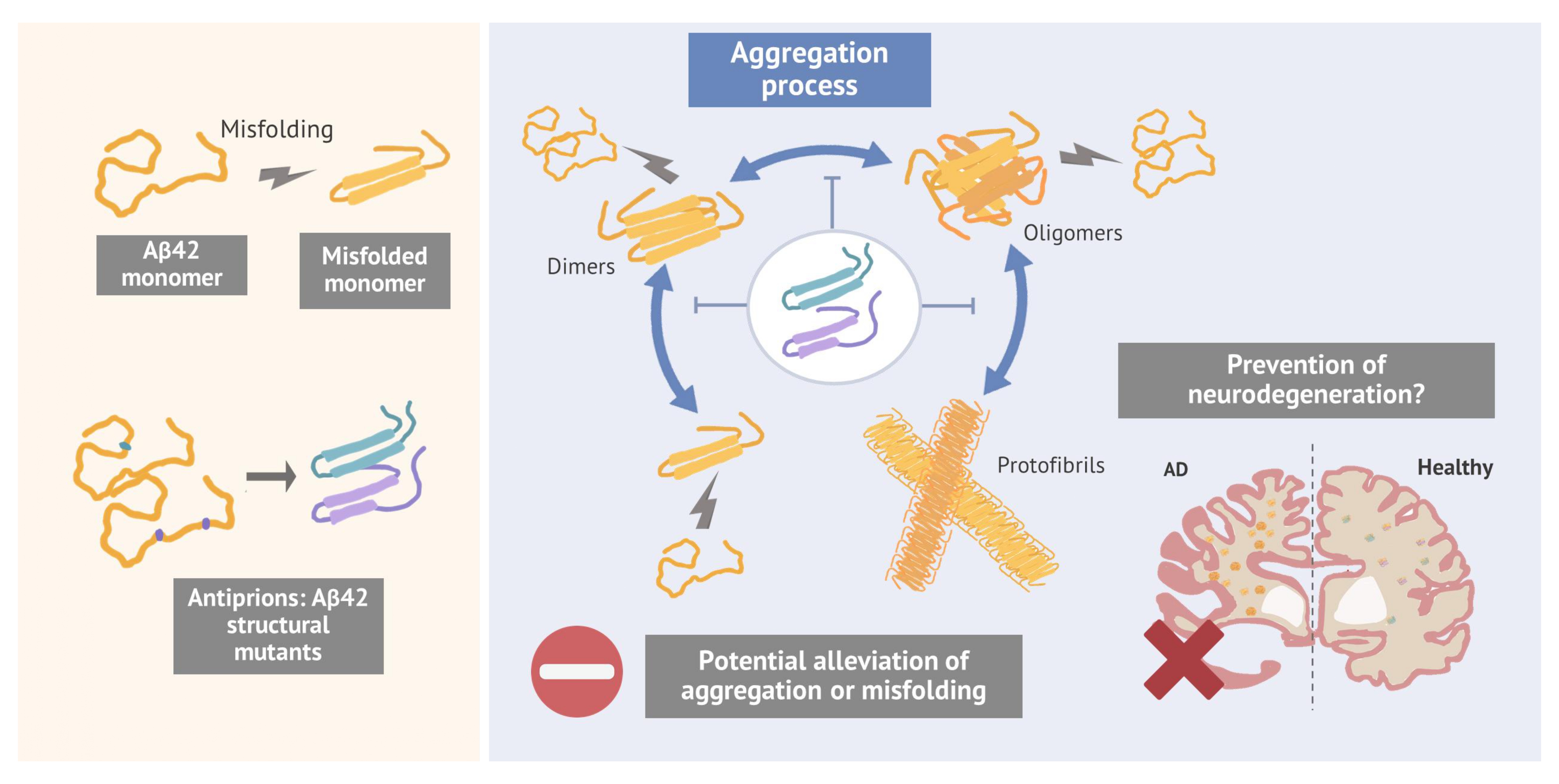
Prions are misfolded protein isoforms that can self-propagate and promote aggregation, playing a central role in neurodegenerative disorders such as Alzheimer’s disease (AD). In AD, Aβ42 peptides exhibit prion-like behaviour, as their misfolded forms transmit aberrant conformations, leading to aggregate formation and amyloid plaque development. However, recent research suggests that intermediate forms, including oligomers and protofibrils, may be the primary culprits in AD pathogenesis. Current treatments show limited efficacy against these smaller aggregates, highlighting the need for alternative strategies. This study proposes the development of antiprions, modified Aβ42 structures capable of binding to aggregates without propagating misfolding, thereby preventing further growth and alleviating toxicity. An unbiased approach combining deep mutational scanning with a high-throughput system in budding yeast was employed, generating and screening over 4.6 million mutants to assess their impact on Aβ42 aggregation. A custom bioinformatics pipeline identified mutations enriched in a counteraggregation selection assay, suggesting potential antiprion candidates. Notably, the discovery of double mutants with epistatic effects revealed unexpected synergy in counteracting aggregation. Some of these double mutants mitigated the toxicity of wild-type (wt) Aβ42 oligomers in viability functional assay using the PC12 cell line as a neuron-like model. Indirect molecular evidence supports interactions between mutant and wt Aβ42 molecules, and preliminary in silico structural analyses suggest that specific mutations could interfere with fibril formation, potentially underlying the observed toxicity rescue. Ongoing transcriptomic studies in neuronal models aim to further explore the neuroprotective effects of these mutants, potentially paving the way for novel therapeutic strategies.